Disease diagnostics using machine learning of immune receptors
Maxim E. Zaslavsky∗, Erin Craig∗, Jackson K. Michuda, Nidhi Sehgal, Nikhil Ram-Mohan, Ji-Yeun Lee, Khoa D. Nguyen, Ramona A. Hoh, Tho D. Pham, Katharina Röltgen, Brandon Lam, Ella S. Parsons, Susan R. Macwana, Wade DeJager, Elizabeth M. Drapeau, Krishna M. Roskin, Charlotte Cunningham-Rundles, M. Anthony Moody, Barton F. Haynes, Jason D. Goldman, James R. Heath, Kari C. Nadeau, Benjamin A. Pinsky, Catherine A. Blish, Scott E. Hensley, Kent Jensen, Everett Meyer, Imelda Balboni, Paul J Utz, Joan T. Merrill, Joel M. Guthridge, Judith A. James, Samuel Yang, Robert Tibshirani, Anshul Kundaje, Scott D. Boyd
Clinical diagnosis typically incorporates physical examination, patient history, and various laboratory tests and imaging studies, but makes limited use of the human system’s own record of antigen exposures encoded by receptors on B cells and T cells. We analyzed immune receptor datasets from 593 individuals to develop MAchine Learning for Immunological Diagnosis (Mal-ID), an interpretive framework to screen for multiple illnesses simultaneously or precisely test for one condition. This approach detects specific infections, autoimmune disorders, vaccine responses, and disease severity differences. Human-interpretable features of the model recapitulate known immune responses to SARS-CoV-2, Influenza, and HIV, highlight antigen-specific receptors, and reveal distinct characteristics of Systemic Lupus Erythematosus and Type-1 Diabetes autoreactivity. This analysis framework has broad potential for scientific and clinical interpretation of human immune responses.
A review of survival stacking: a method to cast survival regression analysis as a classification problem
Erin Craig, Chenyang Zhong, Robert Tibshirani
Right censored data sets are common. To study when or whether an event occurs, we observe subjects over time, and we are rarely able to observe an entire cohort until (1) they have the event of interest or (2) the study is complete.
The standard survival model is the Cox proportional hazards model, which is not always appropriate: it is a linear model that assumes the relationship between the covariates and the hazard is constant through time. There is therefore a need for flexible survival analysis methods.
Many of our favorite methods (boosting, random forests, and deep neural networks) are well-developed for classification and regression, and less so for survival analysis. Further, there are few software packages for survival analysis methods that support common properties of survival data (e.g. time-dependent covariates and truncation).
Our approach, survival stacking, reshapes survival data -- including data with time-dependent covariates and truncation -- so that we can treat survival problems as classification problems. This enables the use of general, flexible classification methods in a survival setting, thereby improving the quality of survival models.
Video credits:
Producers: Erin Craig and Rob Tibshirani
Animation: Jonny Riese
Pretraining and the lasso
Erin Craig, Mert Pilanci, Thomas Le Menestrel, Balasubramanian Narasimhan, Manuel Rivas, Roozbeh Dehghannasiri, Julia Salzman, Jonathan Taylor, Robert Tibshirani

Pretraining is a popular and powerful paradigm in machine learning. As an example, suppose one has a modest-sized dataset of images of cats and dogs, and plans to fit a deep neural network to classify them from the pixel features. With pretraining, we start with a neural network trained on a large corpus of images, consisting of not just cats and dogs but hundreds of other image types. Then we fix all of the network weights except for the top layer (which makes the final classification) and train (or “fine tune”) those weights on our dataset. This often results in dramatically better performance than the network trained solely on our smaller dataset.
In this paper, we ask the question “Can pretraining help the lasso?”. We develop a framework for the lasso in which an overall model is fit to a large set of data, and then fine-tuned to a specific task on a smaller dataset. This latter dataset can be a subset of the original dataset, but does not need to be.
We find that this framework has a wide variety of applications, including stratified models, multinomial targets, multi-response models, conditional average treatment estimation and even gradient boosting. In the stratified model setting, the pretrained lasso pipeline estimates the coefficients common to all groups at the first stage, and then group-specific coefficients at the second “fine-tuning” stage. We show that under appropriate assumptions, the support recovery rate of the common coefficients is superior to that of the usual lasso trained only on individual groups. This separate identification of common and individual coefficients can also be useful for scientific understanding.
MMIL: A novel algorithm for disease associated cell type discovery
Erin Craig∗, Timothy Keyes∗, Jolanda Sarno, Maxim Zaslavsky, Garry Nolan, Kara Davis, Trevor Hastie, Robert Tibshirani
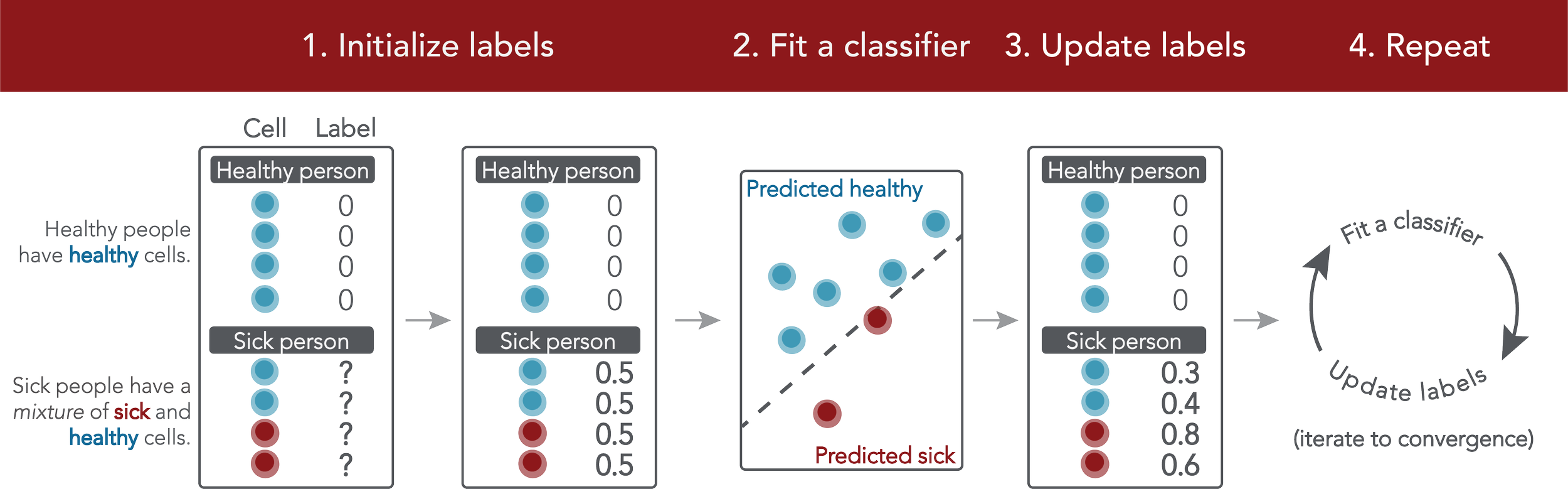
Single-cell datasets often lack individual cell labels, making it challenging to identify cells associated with disease. To address this, we introduce Mixture Modeling for Multiple Instance Learning (MMIL), an expectation maximization method that enables the training of cell-level classifiers using patient-level labels. Our approach can be used to train e.g. lasso logistic regression models, gradient boosted trees, and neural networks. When applied to clinically-annotated, primary patient samples in Acute Myeloid Leukemia (AML) and Acute Lymphoblastic Leukemia (ALL), our method accurately identifies cancer cells, generalizes across tissues and treatment timepoints, and selects biologically relevant features. In addition, MMIL is capable of incorporating cell labels into model training when they are known, providing a powerful framework for leveraging both labeled and unlabeled data simultaneously. Mixture Modeling for MIL offers a novel approach for cell classification, with significant potential to advance disease understanding and management, especially in scenarios with unknown gold-standard labels and high dimensionality.
Finding and assessing treatment effect sweet spots in clinical trial data
Erin Craig, Donald Redelmeier, Robert Tibshirani

Identifying heterogeneous treatment effects (HTEs) in randomized controlled trials is an important step toward understanding and acting on trial results. However, HTEs are often small and difficult to identify, and HTE modeling methods which are very general can suffer from low power.
We present a method that exploits any existing relationship between illness severity and treatment effect, and identifies the "sweet spot", the contiguous range of illness severity where the estimated treatment benefit is maximized. We further compute a bias-corrected estimate of the conditional average treatment effect (CATE) in the sweet spot, and a p-value.
Because we identify a single sweet spot and p-value, our method is straightforward to interpret and actionable: results from our method can inform future clinical trials and help clinicians make personalized treatment recommendations.